Data analysis code
Many of our research projects have involved some sort of “flexible” analysis, meaning that we either developed a new analytical statistical test, developed a new simulation-based test, or conducted custom simulations to test the sensitivity of our results to some of the assumptions we made. Though the details of all these methods and simulations appear in print, I provide here the computer code necessary to carry them out.
I recommend viewing these files within a code-highlighting text editor, such as Textwrangler (for MacOS) or jEdit (for Windows) or in RStudio (either OS). Conceptual background can be found in the referenced documents here, with much more detail usually in the supplementary material. Specific details on implementation are found within the function files themselves, including the necessary input, the returned output, and the format of both. In some cases this also means that the way to estimate the maximum likelihood parameter values and confidence bounds are demonstrated via commented sections within the function files.
***Note that we have published more recent code as part of our supplementary material for papers. Nearly all of that code—which typically includes full code for reproducing all statistical analyses, tables, and figures that appear in each paper—is deposited on Dryad or FigShare.
Misc. phylogenetic functions that I have written for published work
Test: There are three functions in this zipped folder. The first two relate to Ornstein-Uhlenbeck models of character evolution along a phylogeny, which I frequently use for my research on phenotypic evolution. The third I wrote in response to plotting ancestral-state estimates for many models – all the different single-line operations are often fine for one figure, but very tedious if you are making many.
Function 1: Calculating AIC weights. This simply calculates the AIC weights for various models. It’s simple but I did not find something already written in R.
Function 2: Producing an OUCH-format data matrix from a standard data frame and a list of internal ancestral states in the ‘phylo’ format. There are many implementations of the OU model, each with its own strengths. OUCH has many advantages but its phylogeny format is distinct from the most commonly used phylogenetic format in R, from the package ape (class ‘phylo’). What this means is that if you estimate ancestral states using the R packages ape or phytools or diversitree, those estimates will not be in the same order as the internal node labels in ouch. The subsequent manual labeling of internal states that I used to do for ouch was time-consuming, tedious, and error-prone when analyzing large trees or posterior distributions of trees, so I wrote this function. It automates that process by producing an ouch data frame from a list of internal states (of the discrete “selective regime”) in the node order of class ‘phylo’ and a data frame with continuous data and the tip states for the selective regime.
Function 3: Plotting ancestral-state estimates for discrete characters, in the form of a pie chart. This function and its arguments are relatively complicated, but their use is easy to see with examples, which are provided within the function file itself. It should be pretty clear to those that are already familiar with doing this kind of plot.
Computing environment: R
Files: Zipped folder
Tests of convergence and history
Paper: Moen et al. 2016 Syst. Biol. 65:146–160. Supp. Mat.
Test: In this paper we develop a new test of the importance of a lag time for adaptation for explaining phenotypic diversity among species that inhabit the same environment. The files available at Dryad include the R code and two data files necessary to carry out an example analysis (all detailed as comments within the R function).
Computing environment: R
Files: Dryad data repository
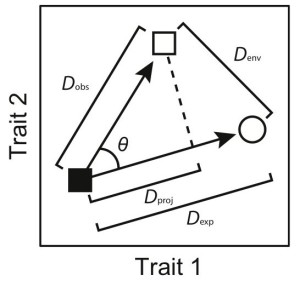 Code for analyses of convergence, conservatism, and multivariate covariance between morphology and performance
Code for analyses of convergence, conservatism, and multivariate covariance between morphology and performance
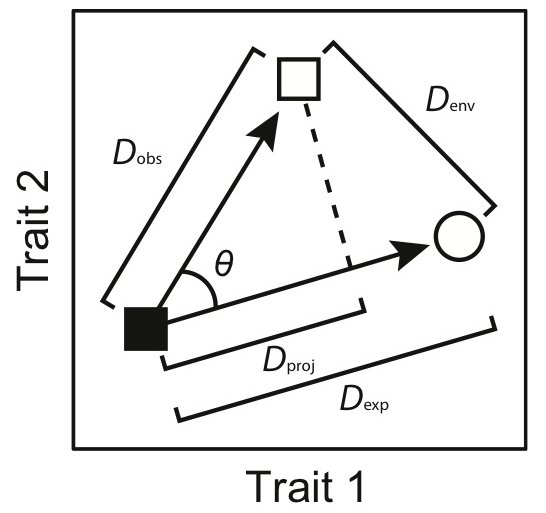
Paper: Moen et al. 2013 Proc. R. Soc. Lond. B 280:20132156. Supp. Mat.
Test: These are various functions I wrote for the above paper, including statistical tests we developed specifically for the paper. The zipped folder also has two functions for carrying out a two-block partial least-squares analysis and associated significance tests for axes (as described by Rohlf and Corti 2000 Syst. Biol. 49:740–753). Full details, including input format and output of the functions, are found within the function files themselves.
Computing environment: R
Files: Zipped folder
Testing whether a regression relationship is stronger than that expected from random sampling
Paper: Wiens et al. 2011 Ecology Letters 14:643–652. Supp. Mat. (starting at line 465)
Test: One of the ideas we tested in this paper was whether the range of body sizes found in a community increased with the species richness of the community. Because the range of an unbounded distribution will necessarily increase with sample size, we wanted to test whether the relationship we found was stronger than expected as an artifact of sampling. Thus, I derived a simple simulation in which we constructed random communities and then estimated the regression between body-size range and community species richness for each of these null communities. This formed the sampling-based null distribution for our empirical regression.
Computing environment: R
Files: Zipped folder
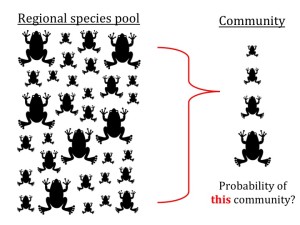 Test of community assembly
Test of community assembly
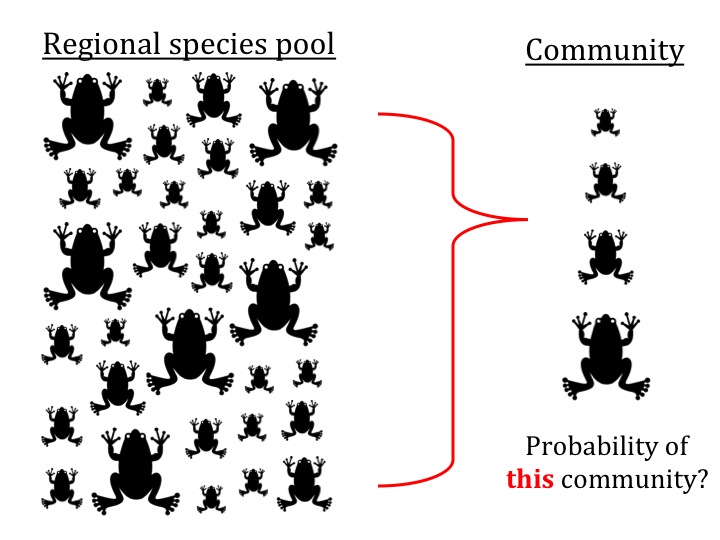
Paper: Moen and Wiens 2009 Evolution 63:195–214. Supp. Mat.
Test: This function is to test whether the phenotypic composition of a community (e.g., in body size) represents a random subset of the source pool, as is often done in community assembly studies. Note that this is an exact likelihood test of what is often approximated through simulation of random community assembly. Our test is based on the hypergeometric probability distribution, a model of sampling without replacement. It allows one to estimate specific “bias” parameters, in effect to estimate which parts of the phenotypic distribution are different in a given community when compared to the source pool.
Computing environment: MatLab
Files: Zipped folder
Test of the importance of in-situ evolution versus ecologically conservative dispersal
Paper: Moen et al. 2009 Evolution 63:3228–3247. Supp. Mat.
Test: In this paper we tested a variety of hypotheses about how phenotypic evolution within a region (on the one hand) and dispersal of species from outside a region (on the other) have affected the phenotypes that we see in communities. One of the tests is simply comparing the relative contribution of these two processes to the phenotypes in a single community. I developed a test to statistically test whether one process was significantly more important than the other. This test is effectively the same as the above test (i.e., based on the hypergeometric probability distribution), but a bit simpler due to only having two categories instead of four. I also conducted simulations to see how the power of this test varied based on the community size.
Computing environment: MatLab
Files: Zipped folder